Aanvragen testen en/of DNA-paspoort
Het DNA zit in iedere cel. Daarom kan de DNA-informatie van enzymen in de lever worden vastgesteld door wat bloed, speeksel of wangslijmvlies te analyseren.
De informatie van het DNA van aantal enzymen bij elkaar vormt een DNA-paspoort voor medicatie (Farmacogenetica Profiel). Het Erasmus MC geeft deze DNA-paspoorten uit.
Hoe aan te vragen?
Via specialist, huisarts of eventueel apotheker. Let op het vermelden van de AGB-code op de aanvraag! De bepalingen kunnen gedaan worden uit bloed of wangslijmvlies. Bij het dichtstbijzijnde bloedafname lab kan bloed afgenomen worden en vervolgens samen met het aanvraagformulier opgestuurd worden naar het Erasmus MC. Voor de afname van wangslijmvlies dient u bij de aanvraag 'Wangslijmvlies (stuur DNA afnamekit naar patient)' aan te vinken, waarna wij de patiënt een DNA-afnamekit toesturen.
Dien hier uw aanvraag digitaal in. (Link openen in Google Chrome)
Het Erasmus MC heeft per 10 januari 2021 een app beschikbaar waarin DNA paspoorten die bij het Erasmus MC zijn gemaakt, onder een code-nummer bekeken kunnen worden op een smartphone.
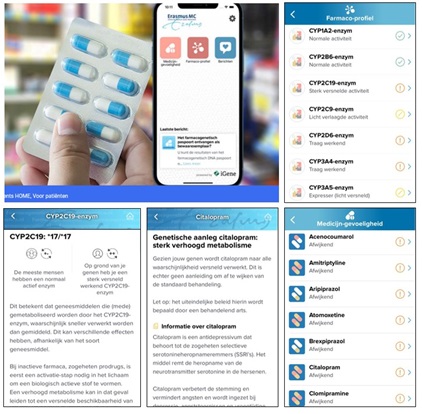
Hierbij is dan ook gelijk alle medicatie zichtbaar in relatie tot uw DNA informatie.
Als u via uw behandelaar een farmacogenetisch profiel laat maken bij het Erasmus MC in Rotterdam, dan kunt u met deze app uw uitslagen direct raadplegen. Zo heeft u uw profiel altijd bij de hand. Daarnaast heeft u het voordeel dat er updates plaatsvinden in het geval er nieuwe medicatie wordt toegevoegd of de doseringsadviezen worden aangescherpt.
U kunt bovendien een DNA rapport opvragen in pdf format, toegestuurd naar uw email adres. Voor patienten die hun paspoort in 2020 bij het Erasmus MC hebben laten maken, is het mogelijk om dit als nog in de App op te nemen. Stuurt u dan een email naar: farmacogenetica@erasmusmc.nl met daarin uw naam, geboortedatum en datum uitgifte Erasmus MC Paspoort. Uw DNA-paspoort gegevens in de App zijn anoniem, en alleen gekoppeld via een uniek codenummer.
De app is te vinden onder “Farmacogenetica Profiel” in de Apple Appstore en in de Android Google Playstore. Download nu de app voor Android of uit de Apple Store.
Farmacogenetica en COVID-19-vaccinatie
De uitslagen op uw DNA-paspoort voor farmacogenetica zijn niet van invloed/geven geen uitsluitsel op eventuele risico’s bij de toediening van de verschillende COVID-19 vaccinaties.
Problemen met aanvragen?
Mocht u problemen hebben met het digitaal aanvragen, dan kunt u hier de printbare versie van het aanvraagformulier downloaden.
Heeft u hulp nodig bij wat aan te vragen voor welk geneesmiddel? Download onderstaand het 'Klinisch relevante enzymen' of 'Substraten' kaartje.
Klinisch relevante enzymen kaartje
Substraten kaartje
Farmacogenetica folder
Wat kunt u ermee?
Wanneer overdosering (toxiciteit) of onderdosering (effectiviteit) ernstige gevolgen kan hebben, kan vooraf bepalen van het farmacogenetisch profiel (screening op relevante genetische polymorfismen) een waardevolle benadering zijn.
Anderzijds kan bij probleemgevallen (patiënten die bij een standaarddosering ongewoon hoge of lage plasmaspiegels hebben) een genetische oorzaak worden aangetoond of uitgesloten. Dit alles om tot meer begrip van een klinisch probleem te komen en een afgewogen keuze voor een alternatief medicijn te kunnen maken.
De landelijke apothekers vereniging KNMP heeft momenteel voor meer dan 80 geneesmiddelen doseringsadviezen beschikbaar op basis van DNA. Iedere apotheker heeft toegang tot die adviezen en kan bewaken dat u altijd medicatie op maat krijgt.
Belangrijk: wijzig als patiënt NOOIT zelf uw medicatie!
Beperkingen Farmacogenetica
De uitslag van de Farmacogenetische test geeft een voorspelling van de enzymactiviteit op basis van erfelijkheid.
Echter, traag metabolisme kan ook voortkomen uit gebruik van co-medicatie. De genetica geeft hierover geen uitsluitsel. Ook worden niet op alle genetische varianten onderzoek gedaan. De kans dat een traag metabolisme door een zeldzame variant wordt veroorzaakt is dus aanwezig, maar is echter niet groot.
Farmacogenetica is een hulpmiddel om het metabolisme op voorhand te voorspellen. Het vervangt niet de noodzaak tot het meten van bloedspiegels of het volgen van biomarkers. Deze laatste methoden zijn echter altijd metingen achteraf.
Achtergond genen en testen
Voor meer achtergrond informatie en interpretatie van de verschillende genen kunnen onderstaande PDF documenten worden gedownload.
Bekijk hier het overzicht van onze wijzingingen in de Farmacogenetica Diagnostiek.
Kosten
Kosten Onderlinge Dienstverlening (Ziekenhuizen, GGZ-instellingen etc.)
De kosten voor de meeste bepalingen zijn €82,50 per test. Voor de enzymen waarvoor meer dan 8 varianten worden getest ligt het tarief hoger. Voor de CYP2D6 ligt het tarief rond de €182,50 en voor de CYP2B6, CYP3A4 en BCHE ligt het tarief rond de €122,50 (NZa 070004 en NZa 070007). (NZa-tarieven aug 2021, prijswijzigingen voorbehouden).
Onderstaand een overzicht van de kosten van de verschillende pakketten:
| Pakket |
Bepalingen |
Kosten |
| DNA Paspoort Basis |
CYP2C9, CYP2C19, CYP2D6, CYP3A4, VKORC1
SLCO1B1 |
€ 624,50 |
| DNA Paspoort Uitgebreid |
CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6,
CYP3A4, CYP3A5, VKORC1, SLCO1B1 |
€ 912,00 |
| Psychiatrie Panel/Mental Health |
CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
€ 542,00 |
| Cardiac Panel |
CYP2C9, CYP2C19, CYP2D6, VKORC1, SLCO1B1,
ABCB1 |
€ 584,50 |
| Pijn Panel |
CYP2C9, CYP2D6, CYP3A4, OPRM1, COMT |
€ 552,50 |
| Oncologie Panel |
CYP2D6, CYP3A4, CYP3A5, DPYD |
€ 470,00 |
Kosten aanvraag via huisarts
Bij aanvraag via de huisarts wordt de rekening rechtstreeks bij uw zorgverzekeraar gedeclareerd. Echter kan dit wel ten laste van het eigen risico gaan.
De kosten voor de bepalingen verschillen per zorgverzekeraar i.v.m. de gemaakte prijs afspraken.
Voor meer informatie betreffende de kosten kunt u contact opnemen met uw zorgverzekeraar. Hiervoor kunt u de NZa 070004 en 070007 opgeven als referentie.
Dekt de verzekering dit?
Als er sprake is van een medische vraag (bijwerkingen, ineffectiviteit) geven de verzekeraars aan dat deze testen in principe vergoed worden. De kosten kunnen echter wel ten laste van het eigen risico gaan. Bij twijfel na te vragen bij de verzekeraar.
Interpretatie van uitslagen
De vertaling van DNA SNP's (single nucleotide polymorphism) naar metabolisme varianten.
Een uitslag wordt bij voorkeur gegeven als de DNA variant die is onderzocht (de DNA variant, oftewel het Single Nucleotide Polymorphism (SNP)), de vertaling naar variant allelen (bijv *1/*4), een indicatie of dit actieve, verminderd actieve of inactieve varianten zijn, en een voorspelling van het metabolisme (traag, intermediair, normaal of ultrasnel).
De DNA varianten zijn met name voor laboratoria zelf van belang, ook om later te kunnen zien wat precies is onderzocht.
Ook is het aantal varianten van belang voor de betrouwbaarheid van de einduitslag. De vertaling naar metabolisme een veralgemenisering: er kan aanzienlijk overlap bestaan tussen deze groepen. Met name tussen intermediaire en normale metaboliseerders. Dit is onder andere afhankelijk van het geneesmiddel. Een CYP2D6 trage metaboliseerder zal voor het antidepressiemiddel imipramine waarschijnlijk het meeste baat hebben bij 30% van de standaarddosering. Terwijl het CYP2D6 geneesmiddel sertraline geen aanpassing behoeft qua dosering, omdat andere enzymen ook en belangrijke rol spelen in de afbraak.
Belangrijk is om te realiseren dat de notatie *1 allen (zijnde het meest voorkomende allel dat codeert voor ene actief enzym) een zogenaamde default waarde is: wanneer er geen varianten worden gevonden, volgt automatisch de uitslag “*1”. De betrouwbaarheid van deze uitslag hangt hier dus af van het aantal onderzochte varianten. En zal nooit 100% zijn.
Hoe wordt Farmacogenetica uitgevoerd?
Dit wordt gedaan met moleculair biologische technieken op DNA dat wordt geïsoleerd uit bloed. Op basis van de testuitslag uit wordt een voorspelling gedaan van de enzymactiviteit.
Omdat niet het gehele DNA wordt onderzocht, bestaat er een kans dat iemand een traag metabolisme heeft door een zeldzame DNA variant. De kans hierop is echter gering.
De waarde van de uitslag wordt mede bepaald door het aantal DNA varianten per enzym dat is onderzocht. Hierdoor lopen ook de prijzen bij de diverse laboratoria wat uiteen. Hoe meer varianten onderzocht zijn, hoe zekerder de uitslag is.
Laboratoria kunnen testen in enkelvoud, in duplo of zelfs met twee verschillende methoden bepalen. Dit is naar inzicht van de laboratoria. De meeste zekerheid wordt verkregen indien twee verschillende methoden worden gebruikt om het DNA profiel vast te stellen. Dit gebeurt onder andere in het Erasmus MC. De op deze website vermelde laboratoria zijn geaccrediteerd en doen mee aan kwaliteitsrondzendingen ten aanzien van Farmacogenetica. Op deze manier wordt de kwaliteit van de analyses gecontroleerd.
De Afdeling Klinische Chemie is ISO-15189 geaccrediteerd onder nummer M098
Laatste nieuws
Farmacogenetica en COVID-19-vaccinatie
De uitslagen op uw DNA-paspoort voor farmacogenetica zijn niet van invloed/geven geen uitsluitsel op eventuele risico’s bij de toediening van de verschillende COVID-19 vaccinaties.
Farmacogenetica App
Per 10-1-2021 heeft het Erasmus MC een app beschikbaar waarin DNA paspoorten die bij het Erasmus MC zijn gemaakt, onder een code-nummer bekeken kunnen worden op een smartphone. Kijk voor meer informatie bij "Aanvragen testen en/of DNA-paspoort".
DPYD genotypering
Per 01-08-2019 heeft het Erasmus MC de *7 bepaling toegevoegd aan de DPYD genotypering, zodat er nu standaard 5 varianten worden getest: *2A, *7, *13, 1236 en 2846. Deze toevoeging is gedaan omdat volgens onze studie het inactieve *7 allel in de Nederlandse populatie vaker blijkt voor te komen (ong 1:100) dan in de algehele populatie is gepubliceerd (1:10.000).
Contact
Voor vragen kunt u contact opnemen met ons secretariaat via 010-7035284 of via de e-mail farmacogenetica@erasmusmc.nl.
Bezoekadres
Erasmus MC
Dr. Molewaterplein 40
3015 GD Rotterdam
Postadres
Erasmus MC
(Inter)Nationaal Expertisecentrum Farmacogenetica
Postbus 2040
3000 CA Rotterdam
