
About Prof. dr. J.N.J. (Sjaak) Philipsen
Erythropoeisis
Hematopoiesis is a self-renewal system that supplies the body's demand for hematopoietic cells throughout an individual's life. Our laboratory has a long standing interest in the development and terminal maturation of erythrocytes, the oxygen-carrying cells in the blood. We are particularly interested in the role of transcription factors and epigenetic regulators in the erythroid developmental program.
Erythropoiesis involves the production of mature enucleated erythrocytes from committed erythroid progenitor cells, which in turn are derived from multi-lineage progenitors and ultimately from the hematopoietic stem cell (HSC). In human the mature erythrocytes turn over at a rate of approximately 1% per day. To maintain the blood count in an adult requires the production of approximately 2.4 x 106 new erythrocytes each second.
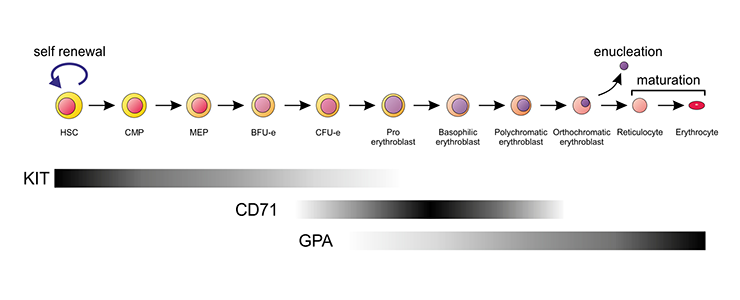
Schematic representation of the erythroid differentiation series in the mouse. Expression of the most commonly used cell surface markers to identify the various stages is indicated by the bars.
Hemoglobin
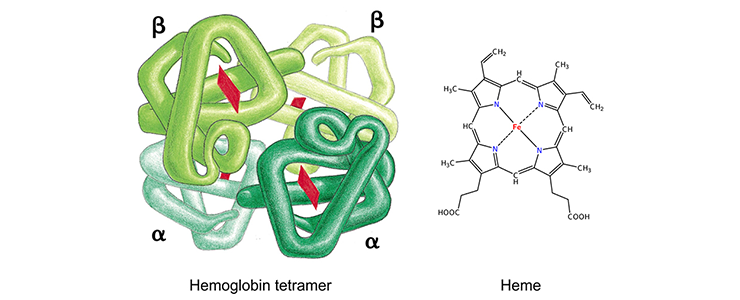
Each globin protein holds an iron-containing heme group (red), which is involved in oxygen binding and release. Figure on the left from Alberts et al, Molecular Biology of the Cell (Garland Science 2008).
Hemoglobinopathies
Balanced production of alpha-like and beta-like globins is important for the formation of hemoglobin tetramers. Insufficient production of alpha-globin leads to alpha-thalassemia, while insufficient production of beta-globin leads to beta-thalassemia. The shortage of functional hemoglobin tetramers, and excess of free globin chains lacking their tetramerization partners, causes a potentially lethal anemia. Hemoglobinopathies can also be caused by pathological globin variants. Substitution of 6th amino acid of beta-globin (glutamic acid for valine, p.E6V) is the most common pathological variant. This is the cause of sickle cell disease.
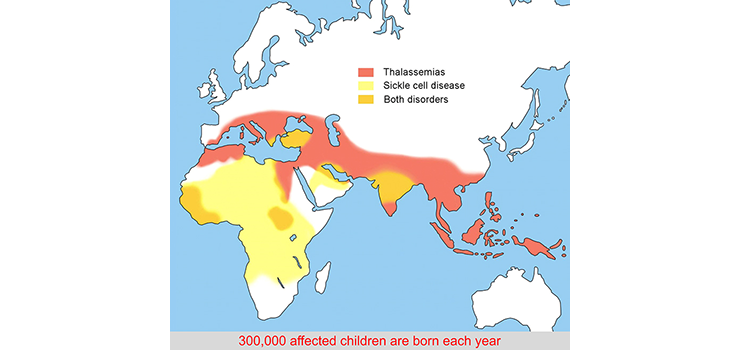
Distribution of hemoglobinopathies
Occurrence of the hemoglobinopathies coincides with areas that are or have been infested with malaria. Heterozygosity provides some protection against the malaria parasite. The carrier frequency in the population world-wide is estimated to be ~5%.
The human α- and ß-globin loci
The α-like globins are encoded by the HBA locus on the tip of human chromosome 16. The ß-like globins are encoded by the HBB locus on human chromosome 11.
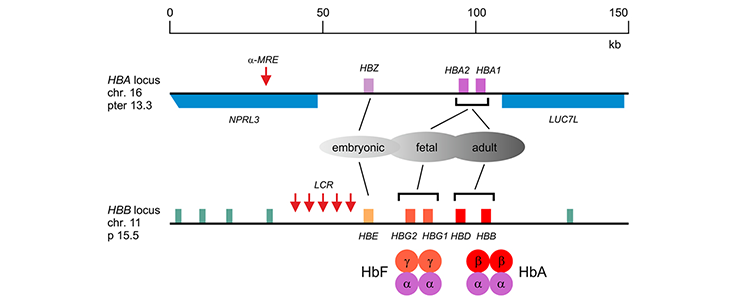
The a-like globin genes (HBZ, HBA1 and HBA2) are flanked by ubiquitously expressed genes (NPRL3 and LUC7L). The major regulatory element (α-MRE) is located upstream of the embryonic zeta-globin gene (HBZ). The ß-like globin genes (HBE, HBG2, HBG1, HBD and HBB) are flanked by putative odorant receptor genes (green). The Locus Control Region (LCR), comprising 5 DNaseI hypersensitive sites required for high-level expression of the b-like globin genes, is located upstream of the embryonic ε-globin gene (HBE). The developmentally regulated expression patterns and the composition of fetal (HbF, α2g2) and adult (HbA, α2ß2) hemoglobin are indicated.
Hemoglobin switching
In humans, expression of the different alpha-like and beta-like globins is developmentally regulated. Distinct hemoglobin tetramers are formed at embryonic, fetal and adult stages. The switch from fetal (HbF, alpha2gamma2) to adult (HbA, alpha2beta2) hemoglobin after birth is clinically relevant. Patients with beta-hemoglobinopathies start displaying disease symptoms during the first year after birth, as expression of HbF gradually tapers off.
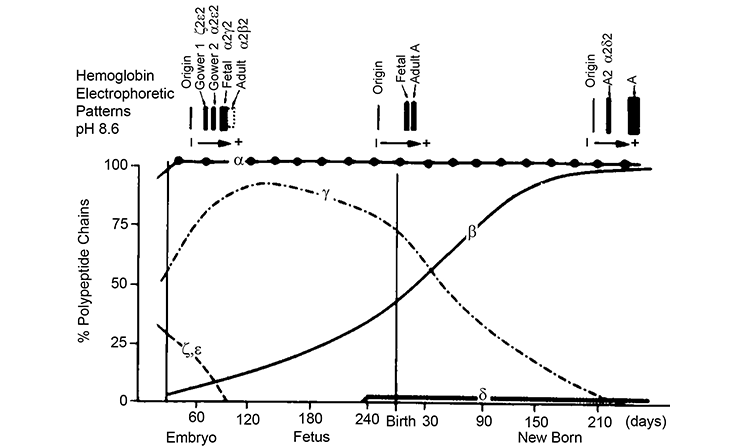
In rare cases, expression of HbF is sustained in beta-hemoglobinopathy patients. Such high HbF levels greatly ameliorate disease symptoms. Reactivation of the HBG1/2 genes in adult erythropoiesis is therefore viewed as an attractive therapeutic approach. To achieve this, we need to understand how the HBG1/2 genes are suppressed in adult erythropoiesis. Intense research efforts by us and other groups have led to the identification of a number of transcription factors that normally suppress the HBG1/2 genes in adult erythroid cells. A major aim of our research projects is to fully characterize the molecular mechanisms that regulate the gamma-globin suppression pathways.
Contact information Philipsen lab
Sjaak Philipsen
e-mail
Visiting address
Dept. of Developmental Biology
Erasmus MC
Faculty building, room Ee-1071b
Dr Molewaterplein 40
3015 GD Rotterdam
The Netherlands
Mail address
Dept. of Developmental Biology
Erasmus MC
Faculty building, room Ee-1071b
PO Box 2040
3000 CA Rotterdam
The Netherlands
The aim of our research projects is to understand the molecular control of erythropoiesis and to fully characterize the mechanisms that regulate the gamma-globin suppression pathways. Below, the KLF1 transcription factor is used as an example to illustrate our research efforts.
KLF1 is an erythroid-specific transcription factor
KLF1 is an erythroid-specific transcription factor, initially discovered by the laboratory of Dr. Jim Bieker in 1992 [PMID: 7682653]. It has a DNA binding domain of three zinc fingers. This DNA binding domain defines it as one of the 27 members of the SP/KLF transcription factor family [PMID: 15820306].

Domains of human KLF1.
Magenta: transactivation domains; 1: aa 1-40; 2: aa 50-90. PEST: sequence rich in proline, glutamic acid, serine, and threonine; 1: aa 32-52; 2: aa 54-67. MIU: motif interacting with ubiquitin; aa 23-36. F: zinc finger domains; 1: aa 281-303; 2: aa 311-233; 3: aa 241-361.
KLF1 is essential for erythropoiesis
Mice lacking KLF1 die around day 14 of embryonic development due to fatal anemia [PMID: 7753194; PMID: 7753195].
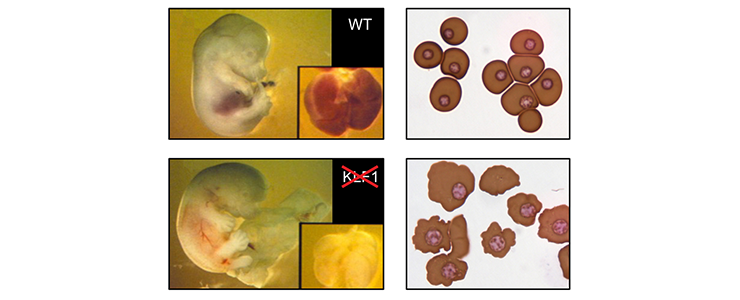
Top left: E13.5 wild-type fetus. Inset: fetal liver. Bottom left: E13.5 KLF1 null fetus, displaying obvious pallor of the fetal liver (inset). Top right: cytospin of E12.5 blood from wild-type fetus, displaying primitive erythrocytes. Bottom right: Cytospin of E12.5 blood from KLF1 null fetus displaying primitive erythrocytes. Note the irregular shape of the cell membranes.
KLF1 has a key role in hemoglobin switching in mice
KLF1 is a specific activator of adult beta-globin expression. In mice carrying a human HBB locus transgene, expression of fetal gamma-globin persists while there is a complete failure to activate b-globin expression [PMID 8918890].
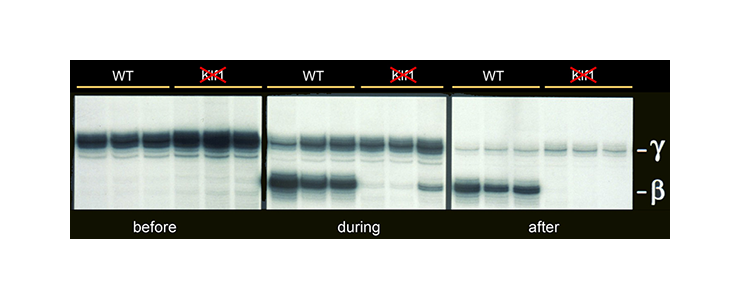
Mice carrying a human HBB locus transgene were crossed with KLF1 null mice. Expression analysis of human gamma- and beta-globin mRNA is shown at various developmental stages - before, during and after hemoglobin switching. In KLF1 null erythroid cells adult beta-globin expression is not activated, whereas expression of fetal gamma-globin persists.
KLF1 has a key role in hemoglobin switching in humans
Our study of a large Maltese pedigree with Hereditary Persistence of Fetal Hemoglobin (HPFH) revealed that haploinsufficiency for KLF1 causes HPFH [PMID: 20676099]. We proposed a model for the role of KLF1 in orchestrating hemoglobin switching which includes our observation that KLF1 activates expression of BCL11A, a direct repressor of the HBG1/2 genes.
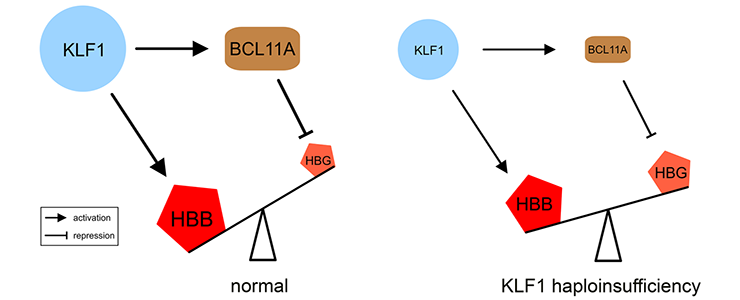
Movie: (left) KLF1 preferentially activates the HBB gene. It also activates the BCL11A gene, and the BCL11A protein silences the HBG1/HBG2 (HBG) genes. (right) Haploinsufficiency reduces KLF1 activity, leading to decreased expression of the BCL11A gene. The diminished amount of BCL11A protein alleviates repression of the HBG1/HBG2 genes. The preferential activation of the HBB gene in adult cells is diminished by reduced KLF1 activity. Collectively, this shifts the balance towards expression of the HBG1/HBG2 genes.
KLF1 variants and erythroid phenotypes in humans
KLF1 variants were thought to be extremely rare causes of human genetic disease. It is now known that a broad range of hitherto unrelated human red cell disorders are caused by variants in KLF1. Remarkably, KLF1 variants reach polymorphic frequencies in populations in which hemoglobinopathies are commonly found. This suggests that KLF1 variants have been under selection and, like the hemoglobinopathies, afford some degree of protection against malaria [PMID: 26903544]. A table of human KLF1 variants reported in the literature can be downloaded here.
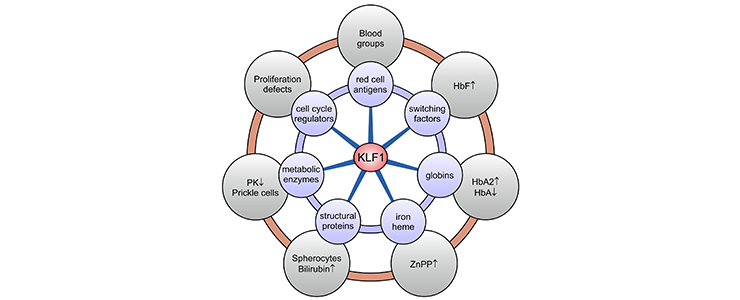
KLF1 target genes and associated clinical phenotypes. KLF1 is a master regulator of ∼700 genes in human erythroid cells involved in a wide variety of molecular processes (blue circles). Deregulated expression of a subset or all of these genes, depending on the KLF1 variant, leads to a diverse array of erythroid phenotypes (gray circles). HbA, adult hemoglobin (alpha2beta2); HbA2, adult hemoglobin 2 (alpha2delta2); PK, pyruvate kinase; ZnPP, zinc protoporphyrin.
PhD students
Promoter (Thesis defence date / Current position)
Steven Heshusius / 02-12-2020 / Investigator at Planbureau for the Environment Alkmaar NL
Silvia Hoeboer / 03-09-2019 / Biology Teacher at Wolfert Bilingual School Rotterdam NL
Maria Mikropoulou / 21-06-2016 / Scientific Marketing Specialist at GenDx NL
Ileana Cantú / 04-11-2015 / Senior Medical Writer at LEO Pharma Copenhagen DK
Divine Kulu / 09-10-2013 / Pharmacy Manager at Walgreens Oklahoma USA
Rejane Hughes Carvalho / 22-02-2012 / Postdoctoral Researcher at FIOCRUZ Salvador BR
Sahar Esteghamat / 20-10-2011 / Director Global Clinical Development at Alexion Boston USA
Pavlos Fanis / 20-10-2011 / Postdoc at The Cyprus Institute of Neurology & Genetics Nicosia CY
Jun Hou / 13-01-2010 / Professor at South China University of Technology Guangdong CN
Co-promoter
Laura Gutiérrez / 09-06-2005 / Head of Research And Development at PlaBiTe Oviedo ES
Pieter Fokko van Loo / 13-04-2005 / VP Research and Development at Gyes Utrecht NL
Rita Ferreira / 27-10-2004 / Research Fellow at Australia National University Canberra AU
Roy Drissen / 27-10-2004 / Senior Researcher at Weatherall Institute of Molecular Medicine Oxford UK
Ulrike Jaegle / 04-06-2003 / VP Global Regulatory Affairs & Preclinical Safety at CureVac Heidelberg DE
Fokke Lindeboom / 02-10-2002 / Clinical Chemist at Franciscus Gasthuis & Vlietland Rotterdam NL
Peter Bouwman / 09-01-2002 / Assistant Professor at LUMC Leiden NL
Gaetano Zafarana / 06-12-2001 / Assistant Professor at The Hospital for Sick Children Toronto CA
Postdocs and technicians
Harmen van de Werken, Head Bioinformatics, Computational Biology, and AI at Erasmus MC Rotterdam NL
Anna Schürmann, Founder Stitch ‘n Bitch Capelle a/d IJssel NL
Teus van Gent, Retired Research Technician
Sylvia Dekker, Research Technician at Technical University Eindhoven NL
Selmar Leeuwenburgh, Research Technician at Erasmus MC Rotterdam NL
Edgar Bonte, Director of Rapido Events BV Amsterdam NL
Bianca den Hamer, Research Technician at Leiden University Medical Center NL
Eva Krpelanova, Medical officer, Classifications and Terminologies Unit, WHO Rotterdam NL
Harald Braun, Postdoc at Flemish Institute for Biotechnology Ghent BE
Hetty Spijker, Nursing home clinician
Robbert Rottier, Professor at Erasmus MC Rotterdam NL
David Whyatt, Associate Professor at University of Western Australia Perth AU
Rita Tewari, Professor at the University of Nottingham UK
Marisol Marin - deceased